Brain structure and function have important implications for depression and its treatment. Several sessions throughout the five-day congress helped to bridge this gap between basic research and clinical practice. Two of these are reviewed below.
Dr. Julio Bobes (Spain) began this session with an introductory presentation during which he described depression as a complex disorder with many different subtypes based on genetic factors (endophenotypes). He showed how these different types of depression are observed in biological markers via neuro-imaging (e.g., the amygdala and prediction response), emotional processing (e.g., facial expression recognition), and genetics. More specifically, Dr. Bobes outlined that depression is associated with hyperactivity in several important areas of the brain. An understanding of these different types of depression and their unique underlying processes can help in setting specific targets for depression treatment.
Dr. Catherine Harmer (U.K.) went on to focus on the impact of depression on emotional processing. She showed that negative affective biases are a key psychological factor in maintaining depression, and described how facial expression recognition can be used to measure emotional bias. Depression, she explained, is associated with increased identification of negative facial expressions and reduced perception of happy facial cues. Of note, she presented data showing that two different antidepressants (citalopram and reboxetine) reduced recognition of negative facial expressions in healthy volunteers and depressed patients, as early as seven days after beginning administration.1 Dr. Harmer also showed how the brain’s response (i.e., in the amygdala) to negative cues is decreased by antidepressants with different mechanisms of action (for example following seven days of treatment with escitalopram). Ultimately, evidence like this suggests that antidepressants affect these neural processes very early in treatment (Figure 1), although the precise mechanisms and comparative effects are not fully known. Regardless, Dr. Harmer went on to show evidence that early emotional processing change (brought about by antidepressant therapy) predicts response vs. non-response, and that early change in amygdala reactivity predicts clinical improvement (for example at week 6 with escitalopram).2 She concluded by pointing out that early changes in emotional processing may help explain the mechanisms of conventional and novel antidepressant drug treatments, and that these effects can also serve as markers of efficacy. Finally, Dr. Harmer asserted that these findings provide a framework in which to explore novel hypotheses in the treatment of depression.
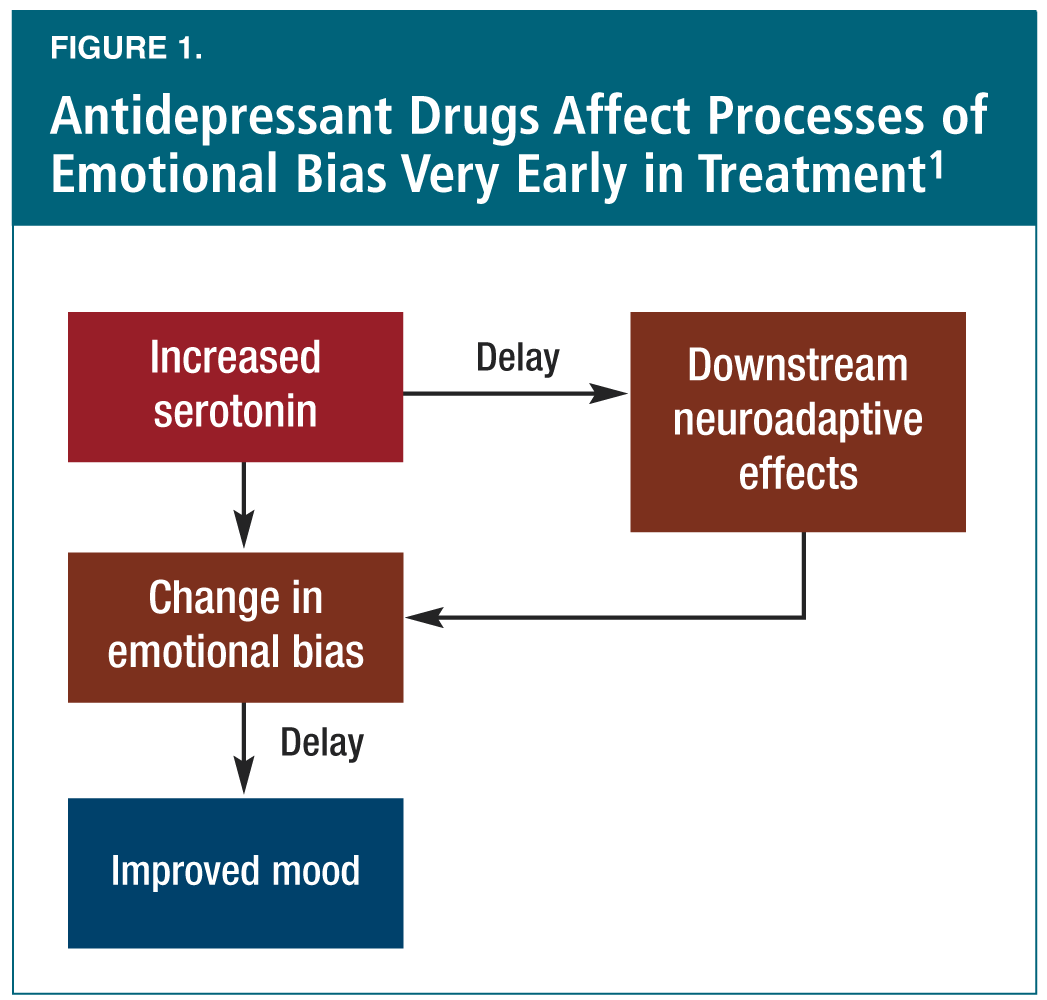
Dr. Philippe Fossati (France) provided a detailed review of the brain effects of antidepressants, describing the “circuit model” of major depressive disorder (MDD).3 He also explained that the variability of brain patterns make these effects of antidepressants difficult to study. These variables, he showed, include clinical/biological heterogeneity (unipolarity vs. bipolarity; the presence or absence of psychiatric comorbidities), actual severity (e.g., MADRS or HAM-D scores; inpatient vs. outpatient populations; mean duration of actual depressive episode), longitudinal severity (first vs. multiple depressive episodes), and functional lesions and adaptive mechanisms. He showed results from a study of the brain effects of agomelatine vs. placebo during a self-referential task in MDD patients (vs. healthy controls),4 and concluded that this agent shows early effects on brain function, targeting emotional processing that may be related to social-functioning recovery.
Finally, Dr. Goran Hajak (Germany) reviewed the clinical implications of restored emotions and improved functioning (mainly through the use of agomelatine). Of interest, he presented a schematic showing that depressed patients, unlike healthy controls, are unable to separate emotional processing from other brain activities such as cognitive and sensorimotor processing.5 This, he proposed, could help explain how the unique actions of agomelatine contribute to improvement across a range of domains and symptoms in depression.
Keeping with the theme of linking brain anatomy and physiology with depression and its treatment, this session began with Dr. Francesc Artigas (Spain) reviewing the brain circuits and targets for cognitive dysfunction in major depression. He described major depression as a disorder of synaptic plasticity6 and of the cortico-limbic circuits. The range of symptoms seen in this disorder include anhedonia, cognitive deficits, depressed mood, somatic changes, fear/anxiety and sleep problems (Table 1). Dr. Artigas showed how the prefrontal cortex (PFC) is the “connector” of the various relevant brain structure functions,7-9 and then provided a detailed review of catecholamines and cognitive function (including the roles of a2-adrenergic receptors and of D1 receptors, and the less-understood roles of several serotonin receptors).10,11 In this context, he showed the in vitro receptor activity and reuptake inhibition of a multi-modal antidepressant currently in development,12,13 and compared the effect of this agent on pyramidal cell firing to that of escitalopram. As he showed, the multi-modal antidepressant (but not escitalopram) dose-dependently increases the discharge rate of specific pyramidal neurons projecting to midbrain, by a mechanism involving 5-HT3 receptors.

Next, Dr. Trevor Robbins (U.K.) briefly reviewed the components of cognition (divided into categories of input, representation, and executive) and the domains of disturbed cognitive function in schizophrenia (speed of processing; attention/vigilance; working memory; verbal learning and memory; visual learning and memory; reasoning and problem-solving; social cognition; positive symptoms).14 He then desribed a series of tests of cognition motivation that can be translated across species (rodents, non-human primates and humans). This translatability, he showed, is cause for “cautious optimism about the use of animal cognition models for [human] drug discovery” relevant to depression and other neuropsychiatric disorders.
Dr. Michael Thase (U.S.) presented the session’s third and final talk, focusing on reviewing the cognitive effects of antidepressants in clinical studies. Dr. Thase began by reviewing the evolution of treatment goals in depression: from a goal of “response” (in which many symptoms remain) in the 1970s, to one of “remission” (in which some symptoms may persist) in the 1990s, and finally to “full functional recovery” (in which symptoms are essentially absent) in the current decade (Table 2).15 He also showed how the common patient descriptors of cognitive symptoms can be mapped to the cognitive domains of attention, memory, psychomotor speed and executive function—and how patients’ feelings of confusion, inadequacy and being overwhelmed overlap onto each of these domains (Figure 2). He reviewed the high prevalence of cognitive dysfunction in depression16 (and as a residual symptom of depression17), and demonstrated that only a small handful of studies have been conducted to examine the effects of current antidepressants on cognitive function. Within this relatively sparse literature, the Rey Auditory Verbal Learning Test (RAVLT) and the Digit Symbol Substitution Test (DSST) have been used to evaluate cognitive performance in MDD.18-20


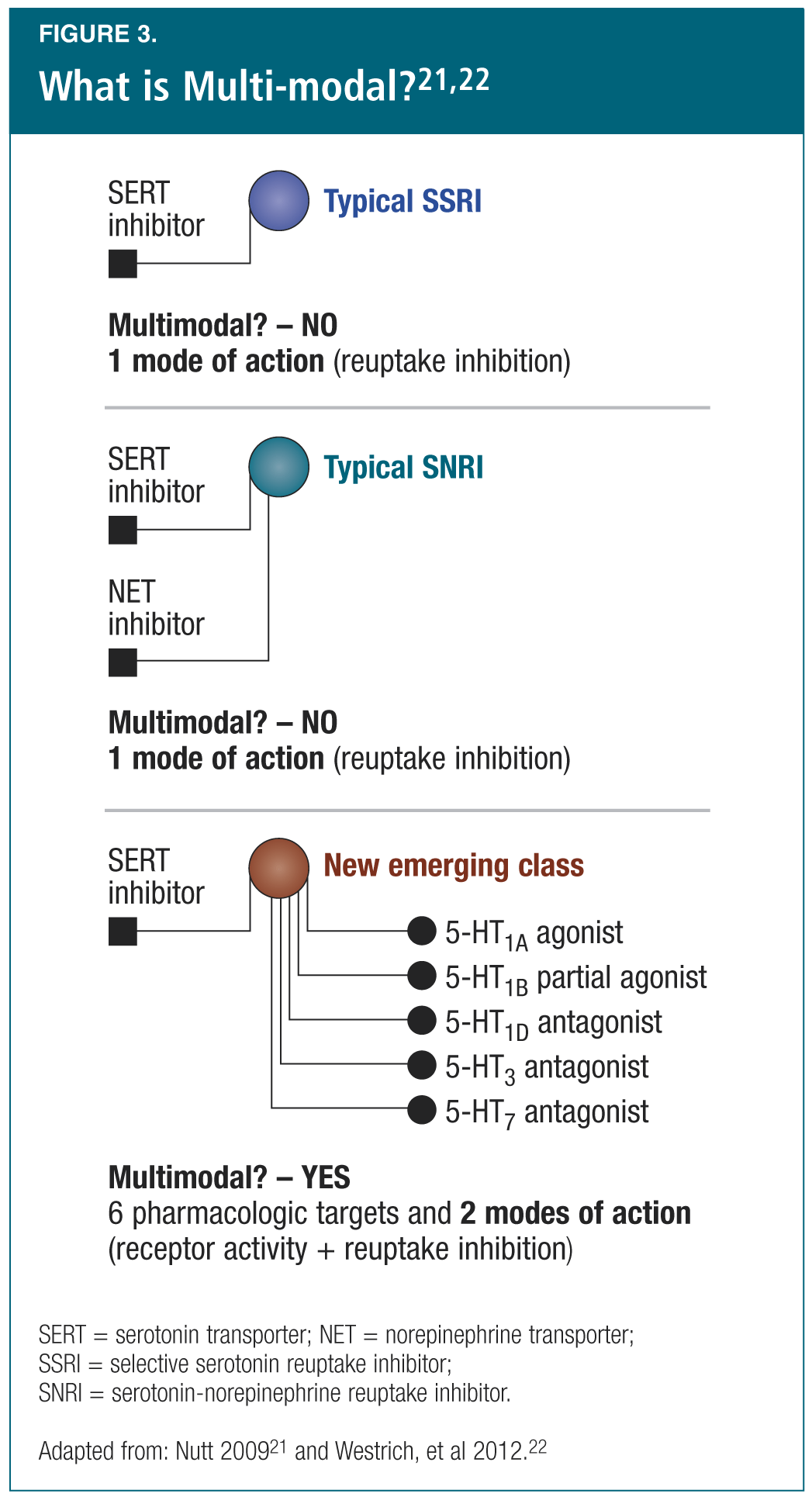
Dr. Thase briefly reviewed the definition of “multi-modal,” describing multi-modal agents as those with multiple pharmacologic targets and multiple modes of action (e.g., receptor activity and reuptake inhibition; Figure 3)21,22 and discussing reasons for which multi-modal compounds may “unite complementary mechanisms of pro-cognitive action” to target cognitive function in depression treatment.23 He concluded by pointing out that ongoing studies are further investigating the effects of multi-modal compounds on the cognitive symptoms of depression.
In addition to sessions focused on basic research like those reviewed above, many other sessions during the ECNP congress were aimed at helping clinicians with their daily patient management. Three of these are reviewed below.
This debate session aimed to answer two key questions faced by clinicians treating depression: 1) what should be the treatment goal (or, what patients want and what doctors think they need); and 2) should treatment be with a single agent or is treatment with two agents better?
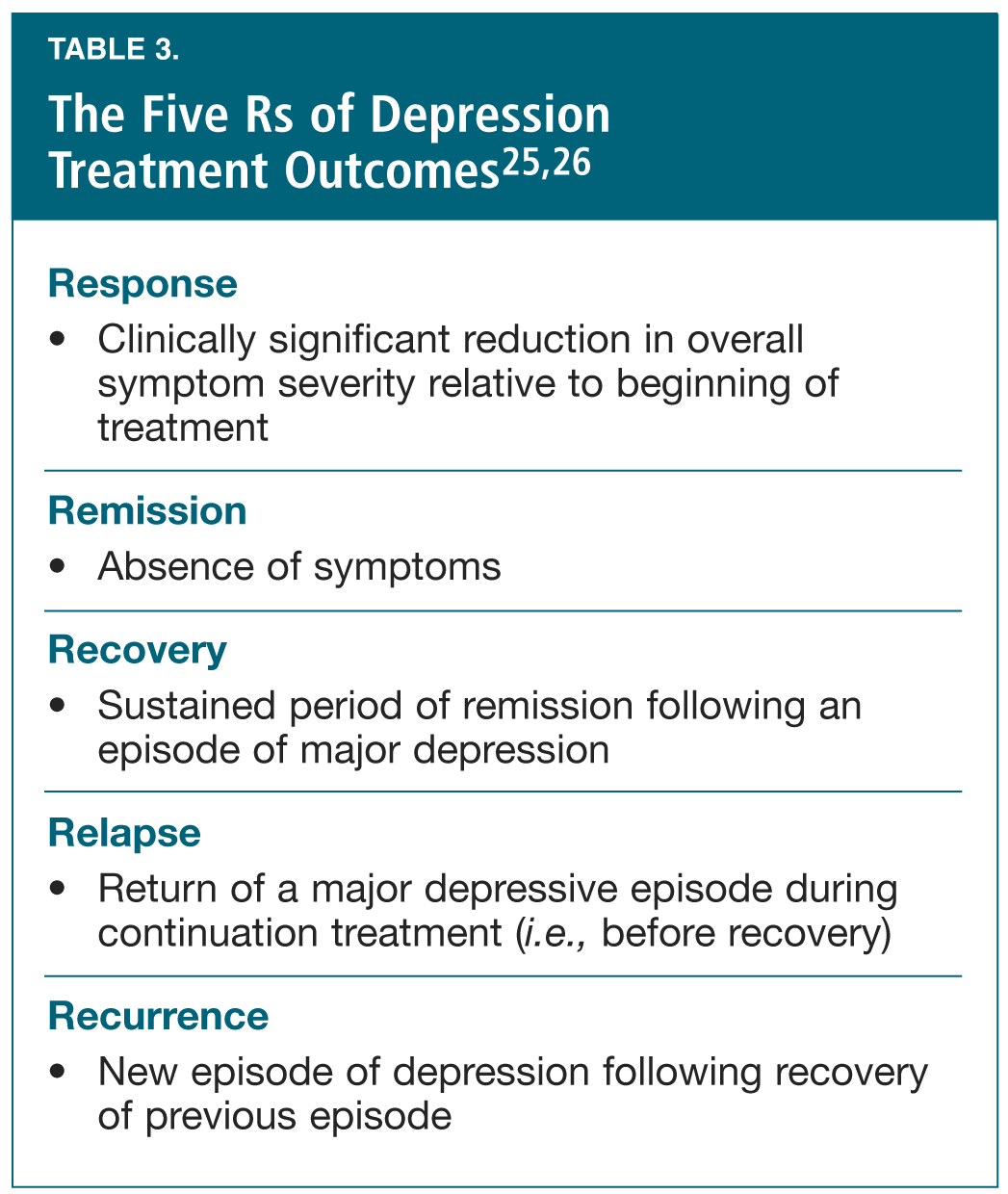
Dr. Charles Nemeroff (U.S.), serving as the session’s chair, provided a brief introduction during which he described the five “Rs” of depression treatment outcome along with their commonly accepted definitions (Table 3).25,26 He also pointed out that, in defining remission and treatment success, one could consider the field of oncology in which 30% remission would surely not be an acceptable goal. In this light, he asserted, the treatment of patients with psychiatric conditions should also aim for higher rates of success.
Dr. Daniel Meron (U.K.) began Part 1 of the debate—focused on treatment goals in depression—by pointing out that the definition of remission is not official or consistent between studies, and suggesting that “functional recovery” should instead be the ideal outcome. He also reviewed data showing that many depressed outpatients (45% in one study he showed) who are in remission according to HAM-D do not consider themselves to be in remission.27,28 Self-described remitters, he showed, report lower levels of depression/anxiety, better quality of life, less functional impairment due to depression, and better coping ability. Meanwhile, a survey of general practitioners and psychiatrists in Belgium29 showed that these two groups of physicians agreed on the criteria for defining cure from depression: they found the SDS and PHQ-9 scales to have the most important content, and focused on interest, mood and suicidal thoughts (and social and occupational functioning included only in the SDS). Finally, Dr. Meron argued for the validity of the CGI scales, showing that CGI-I-defined response corresponded to a 39% reduction in MADRS scores while CGI-S-defined remission corresponded to a MADRS score of 11 (which is defined as remission).30 He concluded that each of the scales remains useful, including the MADRS and HAM-D because of their better inter-rater reliability and allowing for differentiated assessments via subscales.
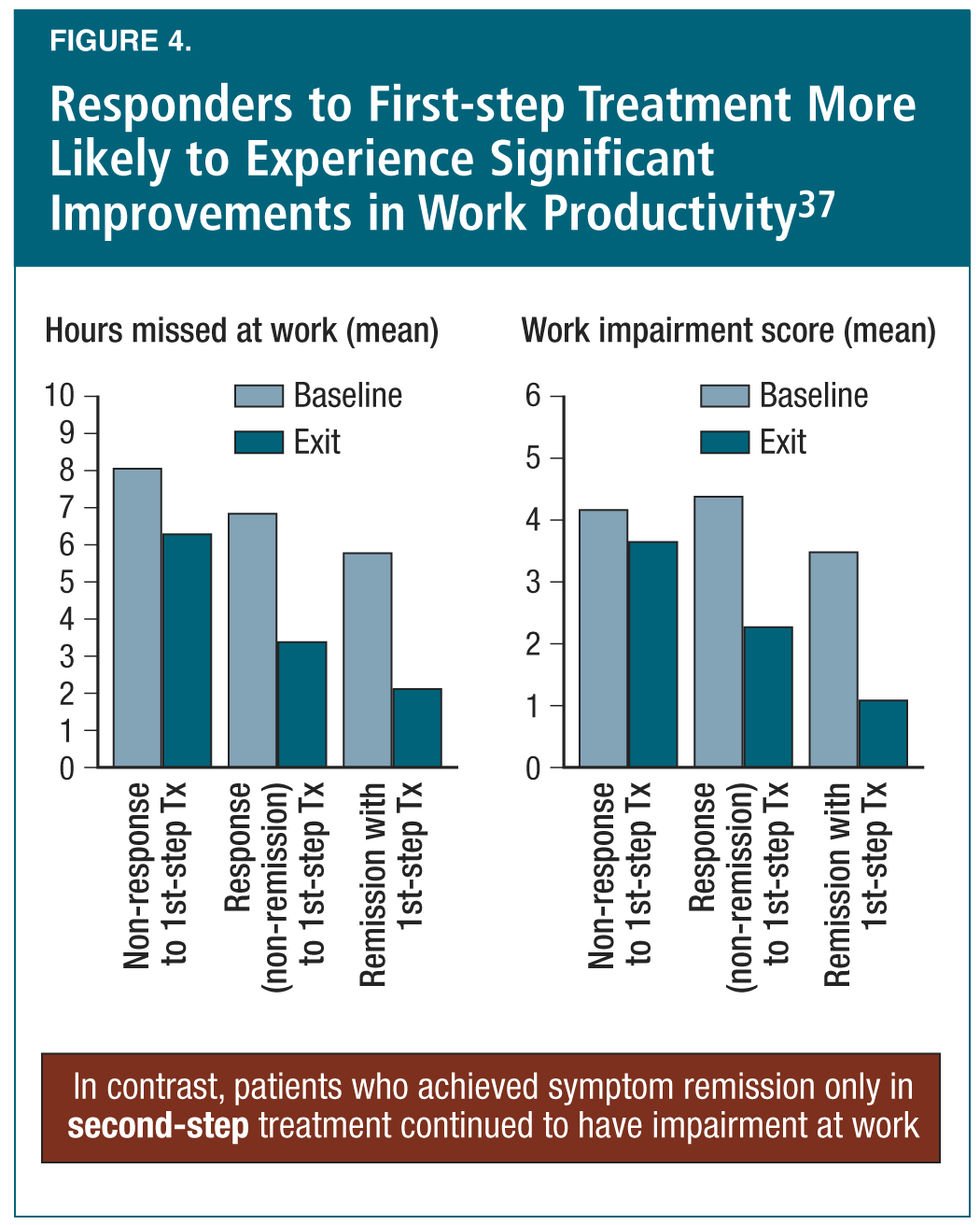
Dr. Roger McIntyre (Canada) countered, showing succinctly that functioning is very important in the patient perspective on depression outcomes, and that patient-reported outcomes predict relapse. He presented the Individual Burden of Illness index for Depression (IBI-D), and showed that this multi-dimensional measure (involving measures of functioning impairment, quality-of-life impairment, and symptom severity) is a more robust predictor of relapse than a unidimensional measure such as the QIDS-SR.31 He also showed that, according to accumulated evidence, symptom remission (“feeling better” with improved symptom scores) does not always translate into functional improvement (“doing better” with improved function scores).32-36 Of particular note, Dr. McIntyre demonstrated the importance of achieving success with the first depression treatment, presenting data that responders to first-step treatment were more likely than non-responders to experience significant improvements in work productivity (while patients who achieved symptom remission only in second-step treatment continued to have impairment at work) (Figure 4).37
Part 2 of the debate led off from this point, with Dr. Meron showing data in favor of combination antidepressant therapy. For example, he showed evidence that initiation of MDD therapy with mirtazapine combined with venlafaxine, bupropion or fluoxetine was more effective than initiation with fluoxetine alone in terms of improving HAM-D score,38 concluding that combination treatments are as well tolerated and more clinically effective, and that use of such combinations from initiation could double the likelihood of remission compared to using a single agent.
Dr. McIntyre challenged this by showing the results of another study, in which three treatment groups (escitalopram monotherapy; escitalopram plus bupropion; venlafaxine plus mirtazapine) did not differ from each other in terms of response or remission in depressed patients.39 He then presented data in favor of a dose-optimization strategy. Focusing on evidence with escitalopram, he showed that dose increase leads to better persistence than switching or adding on agents,40 and that initiation with escitalopram 20 mg was not different from initial combination therapy with bupropion plus escitalopram or with venlafaxine plus mirtazapine in terms of response or remission rates. Furthermore, he presented data from a U.S. claims database showing that an escitalopram dose increase from 10 mg to 20 mg is associated with fewer treament strategy interruptions than switching or adding on therapy, no higher cost than switching therapy, and lower costs than adding on treatment.41
The treatment of patients with depressive disorder and overlapping anxiety disorder/symptoms is another common challenge for clinicians. Setting the stage for addressing this challenge, this session’s co-chair, Dr. David Kupfer (U.S.), summarized the revisions in DSM-5 related to mood and anxiety disorders.42 Elimination of the bereavement exclusion was key among these changes: Dr. Kupfer noted that the exclusion had implied that bereavement typically lasts only two months, when the duration is in fact commonly one to two years. The change to eliminate bereavement as an exclusion criteria will allow clinicians to diagnose depression in bereaved patients when clinically indicated, and therefore facilitate comprehensive treatment. Dr. Kupfer also discussed the addition of the Depression with Anxious Distress diagnosis in order to capture those patients who have significant anxiety associated with their depression but who do not qualify for an anxiety disorder diagnosis. Overall, with its new chapter structure, changes in criteria, new importance of specifiers for more precise assessment, and implications for neuroscience and treatment, Dr. Kupfer asserted that DSM-5 is a “living document” that connects with the biopsychosocial perspective and will allow for more rapid changes as our knowledge base evolves. He also acknowledged that DSM-5 allows for more alignment with the upcoming ICD-11.
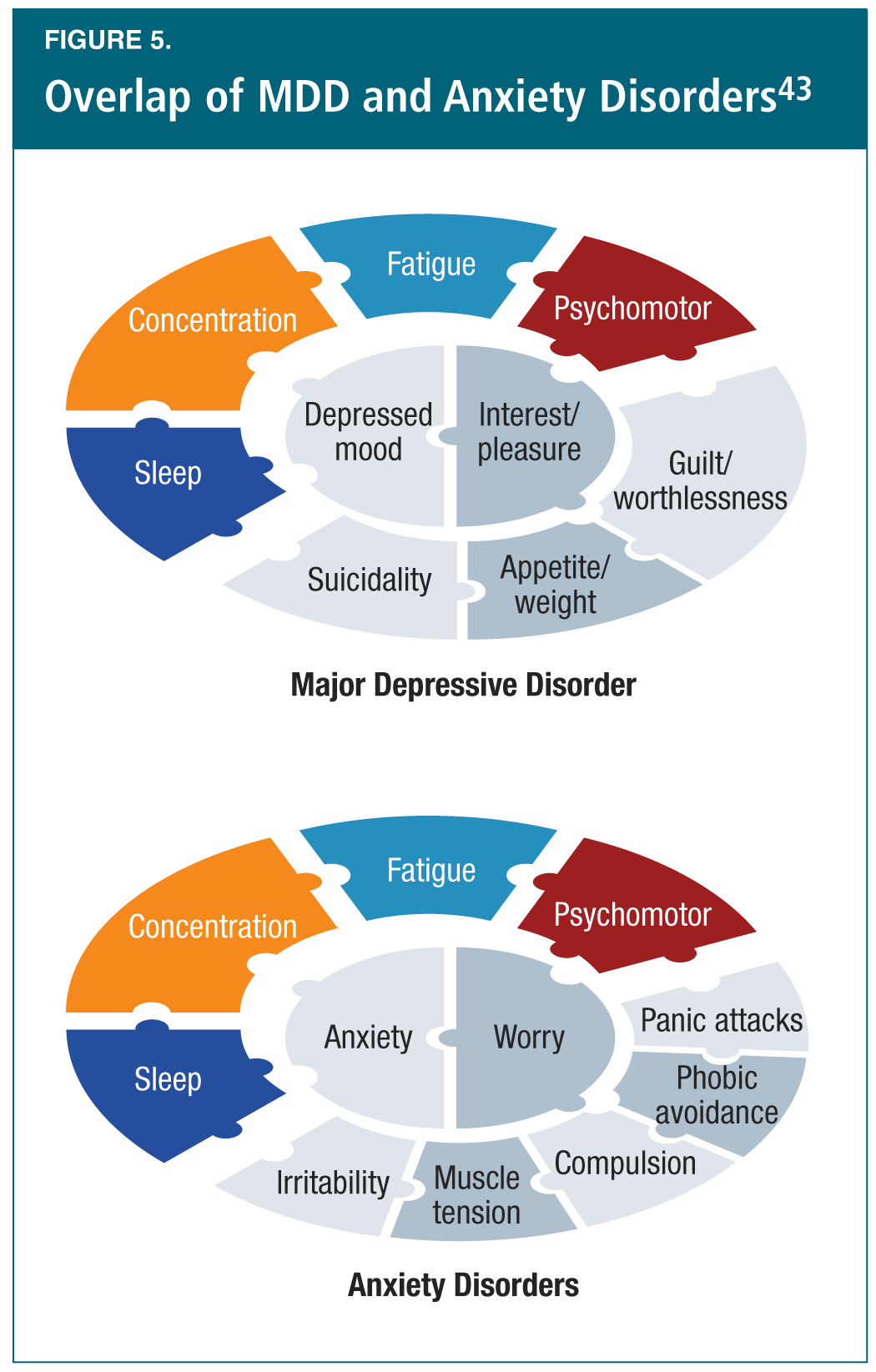
Dr. Stephen Stahl (U.S.) next discussed the biologic and pharmacologic bases for treatment within the anxiety-depression continuum. He began by reviewing the overlapping brain circuitry linked to symptoms of major depression and anxiety disorders, pointing out that the brain has a limited number of “highways” by which it can manifest symptoms (e.g., poor concentration in MDD and in anxiety; Figure 5).43 Dr. Stahl proceeded to “deconstruct” the syndrome of anxiety into its symptoms to illustrate the brain regions and circuits that regulate these symptoms. He also reviewed the role of genetics in the development/emergence of anxiety and depression, describing the differences between “resilient” and “compromised” genotypes, and the effects of various chronic or life stressors on the brain circuits in patients with these genotypes.43 He concluded that neuropharmacologic targets for symptoms of depression and anxiety also overlap, which may explain why some antidepressants can also treat symptoms of anxiety.43
Dr. Koen Demyttenaere (Belgium) concluded this discussion of depression treatment, focusing his talk on examining the “contexts” in which this treatment is understood. He began by discussing some problems with randomized controlled trials, pointing out that clinicians rely heavily on them although they represent only 10-20% of patients, and that placebo response rates have been increasing over time and are correlated with greater numbers of visits to the research clinic.44 He also argued that a publication bias (e.g., 61% of negative studies are not published), reporting bias (e.g., 31% of negative studies are published with a positive “spin”) and an “embracing” bias (e.g., “noisy” vs. “silent” papers) contribute to a problem with the “context” of the available literature.45 Finally, another “contextual” issue which Dr. Demyttenaere addressed is that of what is considered important in treatment, by physicians vs. by patients. In data he showed,46 according to physicians the top priorities include three positive and seven negative affect items; while for patients the top priorities include seven positive and three negative affect items (Table 4). For example, the extent to which life is meaningful was the top issue identified by patients but was at the bottom of the physicians’ list. Moreover, Dr. Demyttenaere pointed out that loss of interest/pleasure is recognized as a core symptom of depression but is not represented on any clinical scales.

This session turned the focus to schizophrenia, and aimed to discuss the important challenge of negative symptoms in this patient group.
Following his introductory remarks in which he outlined the growing interest in negative symptoms in schizophrenia (20% of schizophrenia publications by mid-2012 dealt with negative symptoms, compared to only 5% in 1980), session chair Dr. Josep Maria Haro (Spain) presented a talk prepared by Dr. David Taylor (U.K.) who was unable to attend the congress. As part of this talk, Dr. Haro reviewed the important impact of negative symptoms, pointing out that they frequently cause major burden (for patients, families, etc.)47 even when positive symptoms are controlled, and outlining that the treatment of negative symptoms is an obvious unmet need. He reviewed data showing that some pharmacologic agents have potential in this area, but that current treatments are generally viewed as inadequate by clinicians.
Next, Dr. Pierre-Michel Llorca (France) continued to review the impact of negative symptoms with a focus on patient functioning. He outlined that symptomatic remission is not always associated with sufficient improvement in functioning in all domains;48 that negative symptoms are significantly better predictors of global functioning than all other clinical dimensions;49 that negative symptom severity is the most important factor for prediction of global psychosocial functioning (followed by attention);50 and that negative symptoms were one of the main predictors of the level of burden and distress in a survey of caregivers and patients.51
Dr. Xavier Amador (U.S.) then reviewed the ways in which success has been defined when treating negative symptoms in schizophrenia (the Scale for the Assessment of Negative Symptoms, or SANS), and challenged these conventional clinical expectations and goals by suggesting that the focus should be on loss of motivation (which affects functioning) rather than on the “squeaky wheel” negative symptom of loss of affect. He also reminded the audience of the importance of considering anosognosia in assessing motivation.
Finally, Dr. Philip Tibbo (Canada) provided an overview of (potential) future therapies for the negative symptoms of schizophrenia, and examined the targets and mechanisms of action of these different treatments. He pointed out that efficacy against negative symptoms is not a defining characteristic of atypical antipsychotics,52 and drew attention to an advance in the primarily “dopamine hypothesis” to include the role of GABA (and in particular glutamate) when addressing neurotransmitter dysfunction in schizophrenia.53-55 Finally, Dr. Tibbo reviewed preliminary clinical data with some of the compounds in development aimed at negative symptoms in schizophrenia. While research into many such compounds has been cancelled for various reasons, Dr. Tibbo listed a handful that are still in development (in phase II or III trials).
